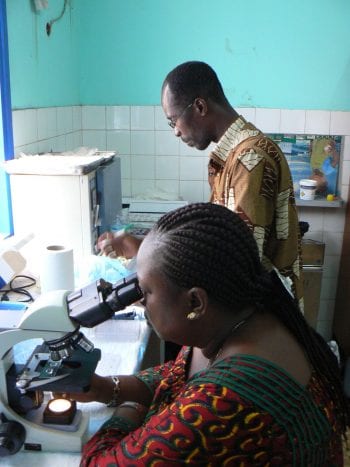
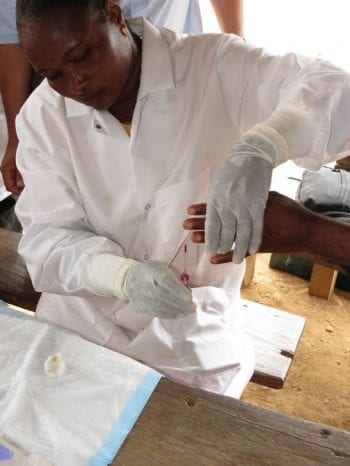
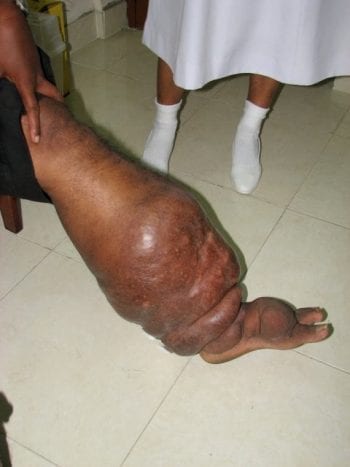
The mission of The DOLF Project is to develop and test improved treatments for onchocerciasis and lymphatic filariasis that will enhance efforts to control and eliminate these important neglected tropical diseases.
NEWS & EVENTS

The DOLF Project has Initiated a Clinical Trial of New Treatments for Onchocerciasis in Liberia
DOLF researchers announce initiation of a major a new clinical trial of treatments for onchocerciasis in Liberia. The study will compare the safety and efficacy of three new combination treatments with a reference treatment of albendazole plus ivermectin. DOLF researchers have worked closely for the past 18 months with colleagues at the National Public Health […]

Gary J. Weil Installed as the Inaugural Gerald and Judith Medoff Professor of Infectious Diseases
Gary J. Weil, MD, co-principal investigator of the DOLF Project, was installed as the inaugural Gerald and Judith Medoff Professor of Infectious Diseases on January 29, 2024 at Washington University in St. Louis.
DOLF Team’s 2023 in Review
Happy 2024 from the DOLF team! From the field to the lab and back again, here is a brief snap shot of our 2023, a year that saw many new advances and developments in our research on lymphatic filariasis (LF) and onchocerciasis (oncho). As an added bonus, we even learned the best bait for catching […]